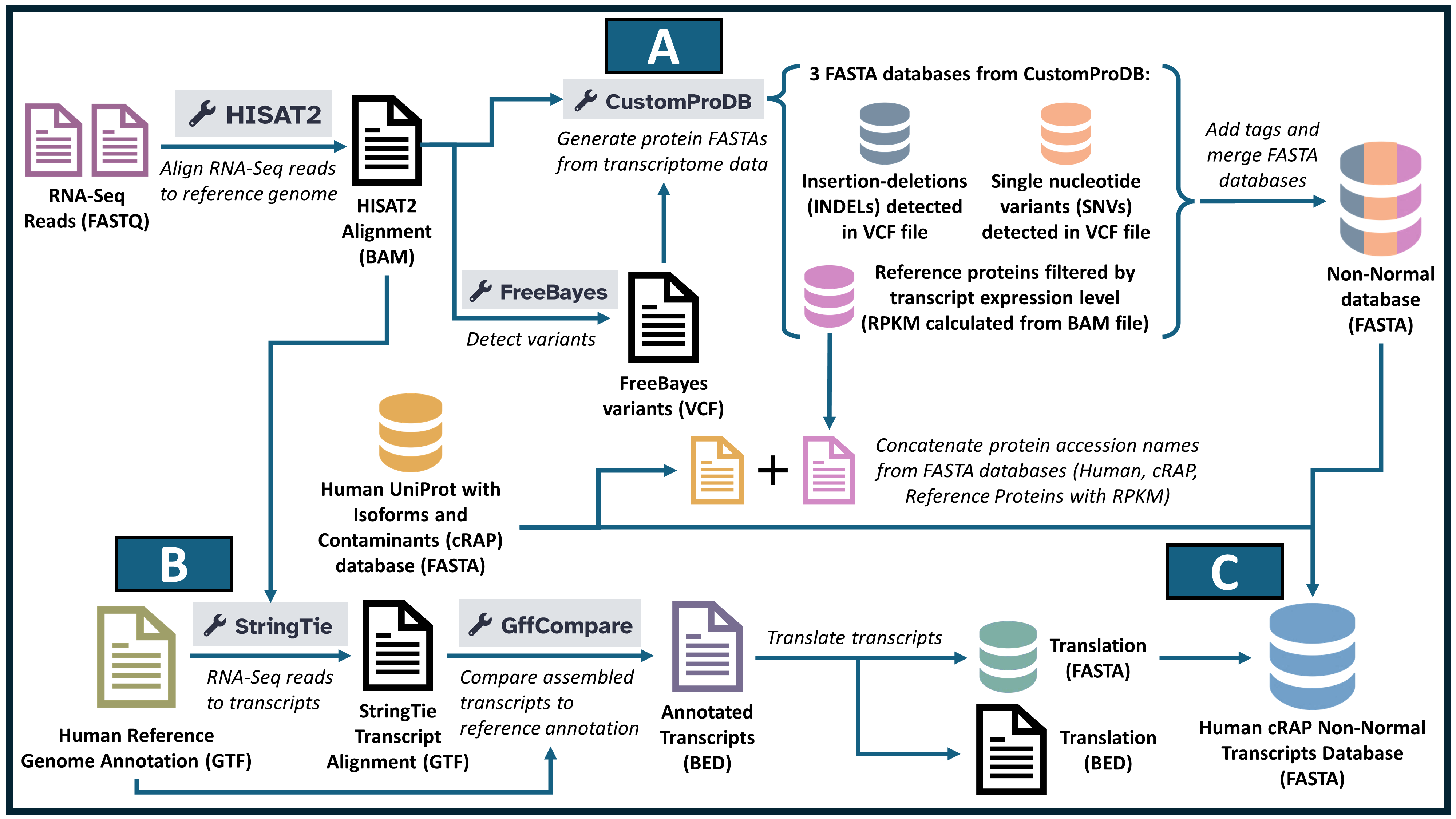
Proteogenomics leverages mass spectrometry (MS)-based proteomics data alongside genomics and transcriptomics data to identify neoantigens—unique peptide sequences arising from tumor-specific mutations. In the initial section of this tutorial, we will construct a customized protein database (FASTA) using RNA-sequencing files (FASTQ) derived from tumor samples. Following this, we will conduct sequence database searches using the resultant FASTA file and MS data to identify peptides corresponding to novel proteoforms, specifically focusing on potential neoantigens. We will then assign genomic coordinates and annotations to these identified peptides and visualize the data, assessing both spectral quality and genomic localization.
In this framework, Proteogenomics incorporates RNA-Seq data to generate tailored protein sequence databases, enabling the identification of protein sequence variants, including neoantigens, through mass spectrometry analysis (Chambers et al. 2017).
In this workflow, (A) Generation of variant database, (B) Generation of assembled protein database, and (C) Merging all databases with known HUMAN protein sequences.
Overview of Non-normal Neoantigen Database Workflow
This tutorial guides users through the process of generating a non-normal variant database. It encompasses essential bioinformatics steps to identify and prepare variant-specific peptides for immunological studies. Below is an overview of each major stage:
Get Data. The workflow begins with uploading raw sequencing data, followed by a quality assessment to ensure data integrity. This step establishes a solid foundation for subsequent analyses by addressing any issues in the initial dataset.
Variant Detection and Mapping. Next, the RNA sequencing data is aligned to a reference genome using tools like HISAT2 and StringTie. Alignment events are detected with specialized tools like Freebayes, CustomProDB, and GFFcompare, which identify non-normal gene transcripts. These tools analyze the resulting alignments to characterize the gene segments in CDS, single nucleotide variants, indels, UTRs, or frameshifts.
Text reformatting and Database generation. Once variants are identified, we generate a customized database and apply various reformatting techniques to tag it, ensuring optimal usability for downstream processing.
Addition of known protein sequences. Known proteomics databases are added to the variant database to create a comprehensive database.
Final Database Construction. The workflow concludes with applying regex adjustments and other formatting functions to standardize the output. This process culminates in creating a comprehensive database of potential non-normal protein sequences, making them ready for experimental validation and clinical exploration.
Get data
Hands-on: Data Upload
Create a new history for this tutorial
Import the files from Zenodo or from
the shared data library (GTN - Material -> proteomics
-> Neoantigen 2: Non-normal-Database-Generation):
Click galaxy-uploadUpload Data at the top of the tool panel
Select galaxy-wf-editPaste/Fetch Data
Paste the link(s) into the text field
Press Start
Close the window
As an alternative to uploading the data from a URL or your computer, the files may also have been made available from a shared data library:
Go into Data (top panel) then Data libraries
Navigate to the correct folder as indicated by your instructor.
On most Galaxies tutorial data will be provided in a folder named GTN - Material –> Topic Name -> Tutorial Name.
Select the desired files
Click on Add to Historygalaxy-dropdown near the top and select as Datasets from the dropdown menu
In the pop-up window, choose
“Select history”: the history you want to import the data to (or create a new one)
Click on Import
Rename the datasets
Check that the datatype
Click on the galaxy-pencilpencil icon for the dataset to edit its attributes
In the central panel, click galaxy-chart-select-dataDatatypes tab on the top
In the galaxy-chart-select-dataAssign Datatype, select datatypes from “New type” dropdown
Tip: you can start typing the datatype into the field to filter the dropdown menu
Click the Save button
Add to each database a tag corresponding to …
Datasets can be tagged. This simplifies the tracking of datasets across the Galaxy interface. Tags can contain any combination of letters or numbers but cannot contain spaces.
To tag a dataset:
Click on the dataset to expand it
Click on Add Tagsgalaxy-tags
Add tag text. Tags starting with # will be automatically propagated to the outputs of tools using this dataset (see below).
Press Enter
Check that the tag appears below the dataset name
Tags beginning with # are special!
They are called Name tags. The unique feature of these tags is that they propagate: if a dataset is labelled with a name tag, all derivatives (children) of this dataset will automatically inherit this tag (see below). The figure below explains why this is so useful. Consider the following analysis (numbers in parenthesis correspond to dataset numbers in the figure below):
a set of forward and reverse reads (datasets 1 and 2) is mapped against a reference using Bowtie2 generating dataset 3;
dataset 3 is used to calculate read coverage using BedTools Genome Coverageseparately for + and - strands. This generates two datasets (4 and 5 for plus and minus, respectively);
datasets 4 and 5 are used as inputs to Macs2 broadCall datasets generating datasets 6 and 8;
datasets 6 and 8 are intersected with coordinates of genes (dataset 9) using BedTools Intersect generating datasets 10 and 11.
Now consider that this analysis is done without name tags. This is shown on the left side of the figure. It is hard to trace which datasets contain “plus” data versus “minus” data. For example, does dataset 10 contain “plus” data or “minus” data? Probably “minus” but are you sure? In the case of a small history like the one shown here, it is possible to trace this manually but as the size of a history grows it will become very challenging.
The right side of the figure shows exactly the same analysis, but using name tags. When the analysis was conducted datasets 4 and 5 were tagged with #plus and #minus, respectively. When they were used as inputs to Macs2 resulting datasets 6 and 8 automatically inherited them and so on… As a result it is straightforward to trace both branches (plus and minus) of this analysis.
Uncompressing data is a crucial first step in many bioinformatics workflows because raw sequencing data files, especially from high-throughput sequencing, are often stored in compressed formats (such as .gz or .zip) to save storage space and facilitate faster data transfer. Compressed files need to be uncompressed to make the data readable and accessible for analysis tools, which generally require the data to be in plain text or other compatible formats. By uncompressing these files, we ensure that downstream applications can efficiently process and analyze the raw sequencing data without compatibility issues related to compression. In this workflow, we do that for both forward and reverse files.
Hands-on: Converting compressed to uncompressed
Convert compressed file to uncompressed. with the following parameters:
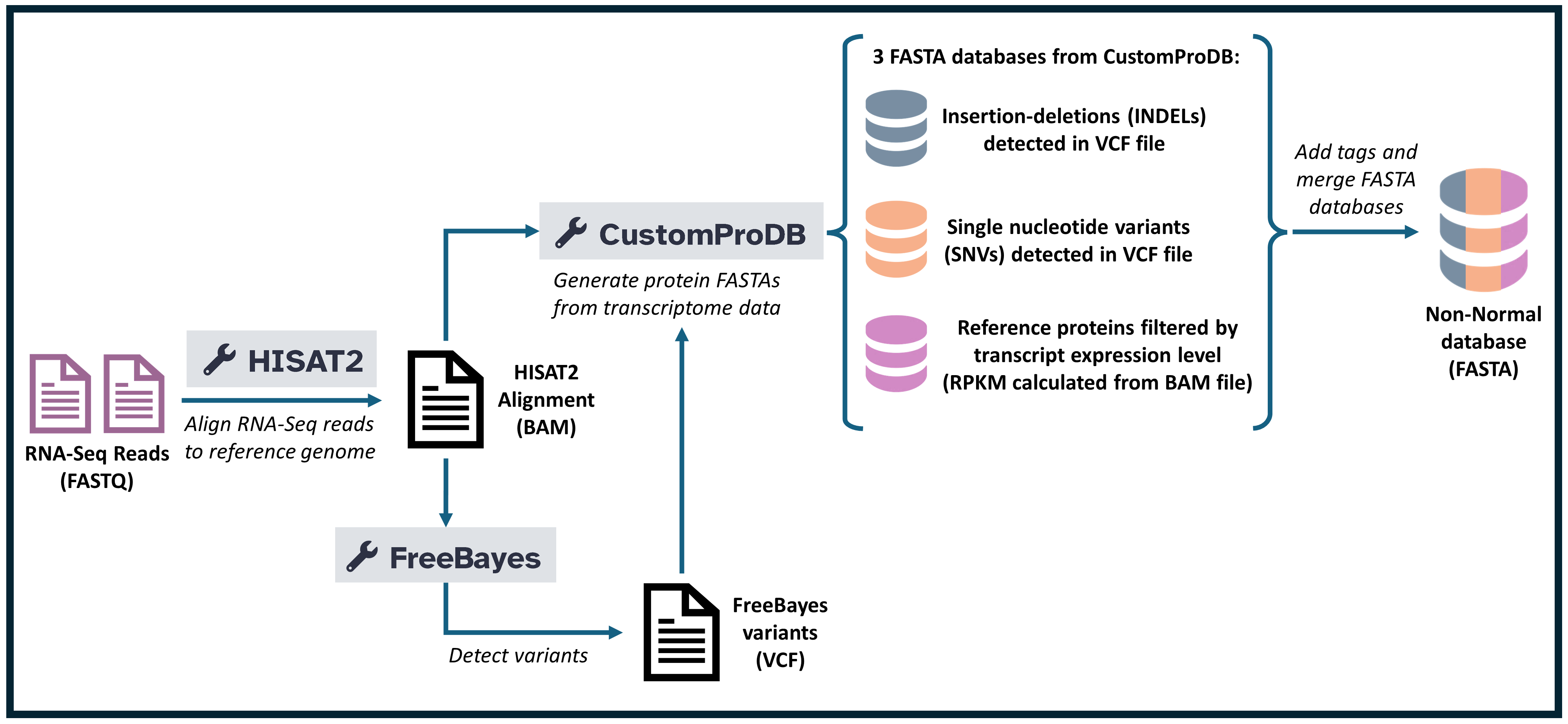
Extracting Single amino acid variants with HISAT and Freebayes
Aligning to the reference genome with HISAT2
HISAT2 is a fast and efficient tool used in bioinformatics workflows to align sequence reads to a reference genome. In this task, HISAT2 is used to align paired-end reads against the human genome version GRCh38 (hg38). This alignment is essential for downstream analyses such as variant calling or transcript quantification. HISAT2 is configured to use default alignment and scoring options to ensure simplicity and speed, which is often suitable for general-purpose analyses.
In this workflow, HISAT2 serves the critical role of mapping raw sequencing data (reads) to a reference genome. This step is a foundation for understanding genetic variation and gene expression in the sample. By aligning the reads to a reference, HISAT2 provides a structured output that can be further analyzed in various bioinformatics applications.
Hands-on: HISAT2
HISAT2 ( Galaxy version 2.2.1+galaxy1) with the following parameters:
“Source for the reference genome”: Use a built-in genome
“Select a reference genome”: Human Dec. 2013 (GRCh38/hg38) (hg38)
“Is this a single or paired library”: Paired-end
param-file“FASTA/Q file #1”: RNA-Seq_Reads_1.fastqsanger (output of Convert compressed file to uncompressed.tool)
param-file“FASTA/Q file #2”: RNA-Seq_Reads_2.fastqsanger (output of Convert compressed file to uncompressed.tool)
“Paired-end options”: Use default values
In “Advanced Options”: Keep all default settings
Question
Why do we need to align sequencing reads to a reference genome in this workflow?
What do paired-end reads represent, and why are they important in this alignment?
Aligning sequencing reads to a reference genome is essential for identifying the location of each read within the genome, which enables analysis of gene expression levels, detection of mutations, and understanding of overall genome structure in the sample.
Paired-end reads are sequences generated from both ends of a DNA fragment, providing more information about the fragment’s size and alignment accuracy. They improve alignment quality and facilitate more accurate mapping, especially for repetitive regions, by providing context from both ends of each read.
Variant Calling with FreeBayes
FreeBayes is a variant calling tool used in bioinformatics to identify genetic variants (such as SNPs, insertions, and deletions) from aligned sequence data. In this step, FreeBayes takes the output from the HISAT2 alignment (BAM or CRAM file) and identifies variations in the aligned reads compared to the human reference genome GRCh38. By running in “Simple diploid calling” mode, FreeBayes is configured to detect variants in diploid samples, assuming two sets of chromosomes as typically found in humans.
In this workflow, FreeBayes performs the essential function of variant calling, which is critical for identifying genetic differences that could be associated with diseases, traits, or other biological characteristics. The output from FreeBayes can then be used for downstream analyses such as functional annotation or association studies.
Hands-on: FreeBayes
FreeBayes ( Galaxy version 1.3.6+galaxy0) with the following parameters:
“Choose the source for the reference genome”: Locally cached
“Run in batch mode?”: Run individually
param-file“BAM or CRAM dataset”: output_alignments (output of HISAT2tool)
“Using reference genome”: Human Dec. 2013 (GRCh38/hg38) (hg38)
“Limit variant calling to a set of regions?”: Do not limit
Why is variant calling important after aligning reads to a reference genome?
What does “Simple diploid calling” mean in the context of FreeBayes?
Variant calling is important because it identifies specific genetic differences between the sample and the reference genome. These differences can provide insights into genetic variation, which may be associated with certain phenotypes, diseases, or treatment responses.
“Simple diploid calling” refers to the assumption that each sample has two copies of each chromosome (diploid). FreeBayes uses this assumption to identify variants present in either one or both copies, which is appropriate for most human genetic analyses.
Customized Database generation using CustomProDB
CustomProDB is a bioinformatics tool used to generate custom protein databases that incorporate specific genetic variants found in a sample. In this task, CustomProDB utilizes genome annotation data (GRCh38/hg38), a BAM file from HISAT2 alignment, and a VCF file from FreeBayes variant calling to create a variant-aware protein database. This database includes the sample’s unique variant proteins, which is essential for personalized proteomics research.
In this workflow, CustomProDB plays a critical role in translating genetic variants identified by FreeBayes into custom protein sequences. This variant-specific database is valuable for applications in proteomics, as it allows for the detection of variant-specific peptides in mass spectrometry data. The generated outputs, including a variant FASTA file and mapping files, support downstream analyses, such as studying how genetic variations may affect protein function or abundance.
Hands-on: CustomProDB
CustomProDB ( Galaxy version 1.22.0) with the following parameters:
“Will you select a genome annotation from your history or use a built-in annotation?”: Use a built-in genome annotation
“Select genome annotation”: Human (Ensembl 78 hsapiens) (hg38/GRCh38)
param-file“BAM file”: output_alignments (output of HISAT2tool)
param-file“VCF file”: output_vcf (output of FreeBayestool)
“Create a variant FASTA for short insertions and deletions”: Yes
“Create SQLite files for mapping proteins to genome and summarizing variant proteins”: Yes
“Create RData file of variant protein-coding sequences”: Yes
Question
Why is it necessary to create a custom protein database with genetic variants?
What is the purpose of creating SQLite and RData files in CustomProDB?
A custom protein database allows for the detection of sample-specific protein variants, which may affect protein structure and function. This is especially useful in personalized medicine, where identifying unique variants helps understand individual differences in disease mechanisms or treatment responses.
The SQLite files map proteins to genome regions and summarize variant proteins, which supports efficient data storage and retrieval for variant analysis. The RData file contains coding sequences for variant proteins, which is useful for further statistical and bioinformatic analyses in R. Although we are not using this in our current workflow, the users can definitely use it to manipulate in their workflows.
Converting FASTA database from CustomPRODB to tabular for text processing with FASTA-to-Tabular
FASTA-to-Tabular is a tool that converts FASTA-formatted sequence files into tabular format, where each sequence entry is represented as a row with separate columns for identifiers and sequences. In this task, FASTA-to-Tabular processes the variant FASTA file generated by CustomProDB to create a tabular representation of the protein variants. This format is often easier to analyze or integrate into downstream data workflows.
In this workflow, FASTA-to-Tabular enables the conversion of variant protein sequences into a structured tabular format, which is helpful for subsequent data processing and analysis. This format allows researchers to efficiently filter, sort, or query specific sequence information and simplifies integration with other data analysis tools or databases. We do this for the indels, single nucleotide variants and rpkm databases.
Hands-on: INDEL - FASTA-to-Tabular
FASTA-to-Tabular ( Galaxy version 1.1.1) with the following parameters:
param-file“Convert these sequences”: output_indel (output of CustomProDBtool)
Hands-on: SNV - FASTA-to-Tabular
FASTA-to-Tabular ( Galaxy version 1.1.1) with the following parameters:
param-file“Convert these sequences”: output_snv (output of CustomProDBtool)
Hands-on: RPKM - FASTA-to-Tabular
FASTA-to-Tabular ( Galaxy version 1.1.1) with the following parameters:
param-file“Convert these sequences”: output_rpkm (output of CustomProDBtool)
Question
Why is it helpful to convert FASTA sequences to a tabular format?
What information is typically retained when converting a FASTA file to tabular format?
Converting FASTA sequences to tabular format makes it easier to visualize, filter, and manage the data. The tabular structure allows researchers to perform sorting, searching, and data manipulation more efficiently, which is beneficial for downstream analysis.
The conversion usually retains the sequence identifiers and the sequences themselves. Each sequence entry in the FASTA file is represented in two columns: one for the identifier and another for the actual sequence, enabling easy access to both key elements of the data.
Manipulating the headers with Column Regex Find And Replace
Column Regex Find And Replace is a tool that applies regular expression (regex) patterns to specified columns in a tabular dataset to find and replace text patterns. In this task, the tool is used on the output from the FASTA-to-Tabular step to standardize or format specific patterns in the data. By applying regex patterns to column c1, the tool identifies and modifies specific sequence identifiers or annotations, making them easier to interpret or use in further analyses.
In this workflow, Column Regex Find And Replace cleans and formats the data in a way that makes identifiers or variant descriptions consistent. This is important for data compatibility, especially when the data needs to be used across different tools or integrated into larger datasets. It ensures that all sequence labels or variant annotations follow a uniform format, which reduces errors in downstream analyses.
Regular expressions are a standardized way of describing patterns in textual data. They can be extremely useful for tasks such as finding and replacing data. They can be a bit tricky to master, but learning even just a few of the basics can help you get the most out of Galaxy.
Finding
Below are just a few examples of basic expressions:
Regular expression
Matches
abc
an occurrence of abc within your data
(abc|def)
abcordef
[abc]
a single character which is either a, b, or c
[^abc]
a character that is NOT a, b, nor c
[a-z]
any lowercase letter
[a-zA-Z]
any letter (upper or lower case)
[0-9]
numbers 0-9
\d
any digit (same as [0-9])
\D
any non-digit character
\w
any alphanumeric character
\W
any non-alphanumeric character
\s
any whitespace
\S
any non-whitespace character
.
any character
\.
{x,y}
between x and y repetitions
^
the beginning of the line
$
the end of the line
Note: you see that characters such as *, ?, ., + etc have a special meaning in a regular expression. If you want to match on those characters, you can escape them with a backslash. So \? matches the question mark character exactly.
Examples
Regular expression
matches
\d{4}
4 digits (e.g. a year)
chr\d{1,2}
chr followed by 1 or 2 digits
.*abc$
anything with abc at the end of the line
^$
empty line
^>.*
Line starting with > (e.g. Fasta header)
^[^>].*
Line not starting with > (e.g. Fasta sequence)
Replacing
Sometimes you need to capture the exact value you matched on, in order to use it in your replacement, we do this using capture groups (...), which we can refer to using \1, \2 etc for the first and second captured values. If you want to refer to the whole match, use &.
Regular expression
Input
Captures
chr(\d{1,2})
chr14
\1 = 14
(\d{2}) July (\d{4})
24 July 1984
\1 = 24, \2 = 1984
An expression like s/find/replacement/g indicates a replacement expression, this will search (s) for any occurrence of find, and replace it with replacement. It will do this globally (g) which means it doesn’t stop after the first match.
Example: s/chr(\d{1,2})/CHR\1/g will replace chr14 with CHR14 etc.
You can also use replacement modifier such as convert to lower case \L or upper case \U. Example: s/.*/\U&/g will convert the whole text to upper case.
Note: In Galaxy, you are often asked to provide the find and replacement expressions separately, so you don’t have to use the s/../../g structure.
There is a lot more you can do with regular expressions, and there are a few different flavours in different tools/programming languages, but these are the most important basics that will already allow you to do many of the tasks you might need in your analysis.
Tip:RegexOne is a nice interactive tutorial to learn the basics of regular expressions.
Tip:Regex101.com is a great resource for interactively testing and constructing your regular expressions, it even provides an explanation of a regular expression if you provide one.
Tip:Cyrilex is a visual regular expression tester.
Hands-on: INDEL-Column Regex Find And Replace
Column Regex Find And Replace ( Galaxy version 1.0.3) with the following parameters:
param-file“Select cells from”: output (output of FASTA-to-Tabulartool)
Why use regular expressions to modify text in a column?
What is the purpose of the generic|INDEL_\1 replacement pattern ( or any other pattern) in this tool?
Regular expressions allow for flexible, precise pattern matching, enabling the user to find and replace complex text patterns within a column. This is useful for standardizing data formats, extracting relevant portions, or appending additional information to entries.
The generic|INDEL_\1 replacement pattern prepends generic|INDEL_ to each entry in the column, standardizing the label format and clarifying that each entry is related to an insertion or deletion (INDEL) variant. This makes it clear in downstream analysis that the sequence is variant-specific.
Converting the manipulated tabular files to FASTA with Tabular-to-FASTA
Tabular-to-FASTA is a tool that converts tabular data back into FASTA format, where each entry has a title and a sequence. In this step, the tool uses the processed output from the Column Regex Find And Replace step to create a FASTA file. Column c1 is set as the title (identifier) for each sequence, and column c2 contains the sequence data. This conversion is useful when the data needs to be returned to a standard sequence format for further bioinformatics analyses.
In this workflow, Tabular-to-FASTA converts the formatted tabular data back into a FASTA file, making it compatible with tools that require FASTA input for further analysis. This step enables the standardized, cleaned sequences from previous steps to be utilized in additional bioinformatics workflows or databases, maintaining the variant-specific information in a commonly used format. We do this for all the tabular files (SNV, INDEL, and RPKM).
Hands-on: INDEL-Tabular-to-FASTA
Tabular-to-FASTA ( Galaxy version 1.1.1) with the following parameters:
param-file“Tab-delimited file”: out_file1 (output of Column Regex Find And Replacetool)
“Title column(s)”: c['1']
“Sequence column”: c2
Rename as Indel-FASTA
Hands-on: SNV-Tabular-to-FASTA
Tabular-to-FASTA ( Galaxy version 1.1.1) with the following parameters:
param-file“Tab-delimited file”: out_file1 (output of Column Regex Find And Replacetool)
“Title column(s)”: c['1']
“Sequence column”: c2
Rename as SNV-FASTA
Hands-on: RPKM-Tabular-to-FASTA
Tabular-to-FASTA ( Galaxy version 1.1.1) with the following parameters:
param-file“Tab-delimited file”: out_file1 (output of Column Regex Find And Replacetool)
“Title column(s)”: c['1']
“Sequence column”: c2
Rename as RPKM-FASTA
Question
Why is it important to convert tabular data back to FASTA format?
What information is typically included in the title and sequence columns in a FASTA file?
Converting tabular data back to FASTA format makes it compatible with a wide range of bioinformatics tools that require FASTA input. It allows the sequence data, now standardized and labeled, to be used in additional analyses, such as protein structure prediction or functional annotation.
The title column usually contains a unique identifier or label for each sequence, often including relevant annotations. The sequence column contains the actual DNA, RNA, or protein sequence. Together, these provide essential details for recognizing and analyzing each sequence entry in downstream applications.
Merging Single amino acid variant databases with FASTA Merge Files and Filter Unique Sequences
FASTA Merge Files and Filter Unique Sequences is a tool that combines multiple FASTA files into a single file and removes any duplicate sequences, keeping unique entries. In this task, the tool takes the FASTA file generated from the Tabular-to-FASTA step and merges it with any other FASTA files in the input list. The tool then filters the sequences to ensure that unique sequences are retained in the final output, which is important for reducing redundancy in the dataset.
In this workflow, FASTA Merge Files and Filter Unique Sequences consolidate all sequence data into a single, non-redundant FASTA file. This step is essential for removing duplicate sequences, which helps streamline the dataset for further analysis. A unique sequence file reduces computational load and minimizes potential biases in downstream applications that could be affected by redundant data. We are merging the indel, snv, and rpkm databases in this step.
Hands-on: FASTA Merge Files and Filter Unique Sequences
FASTA Merge Files and Filter Unique Sequences ( Galaxy version 1.2.0) with the following parameters:
“Run in batch mode?”: Merge individual FASTAs (output collection if the input is a collection)
In “Input FASTA File(s)”:
param-repeat“Insert Input FASTA File(s)”
param-file“FASTA File”: SNV-FASTA (output of Tabular-to-FASTAtool)
In “Input FASTA File(s)”:
param-repeat“Insert Input FASTA File(s)”
param-file“FASTA File”: INDEL-FASTA (output of Tabular-to-FASTAtool)
In “Input FASTA File(s)”:
param-repeat“Insert Input FASTA File(s)”
param-file“FASTA File”: RPKM-FASTA (output of Tabular-to-FASTAtool)>
Rename as Non-normal_CustomProDB_FASTA
Question
Why is it useful to filter out duplicate sequences from a FASTA file?
What does “Run in batch mode” mean in the context of merging FASTA files?
Filtering out duplicate sequences ensures that unique sequences are included, which reduces redundancy in the dataset. This can enhance computational efficiency, prevent overrepresentation of certain sequences, and ensure that analyses are based on a unique set of entries.
In this context, “Run in batch mode” means merging multiple FASTA files into a single collection, even if the input consists of multiple individual FASTA files. This mode consolidates the files to make them easier to manage and analyze as a single dataset.
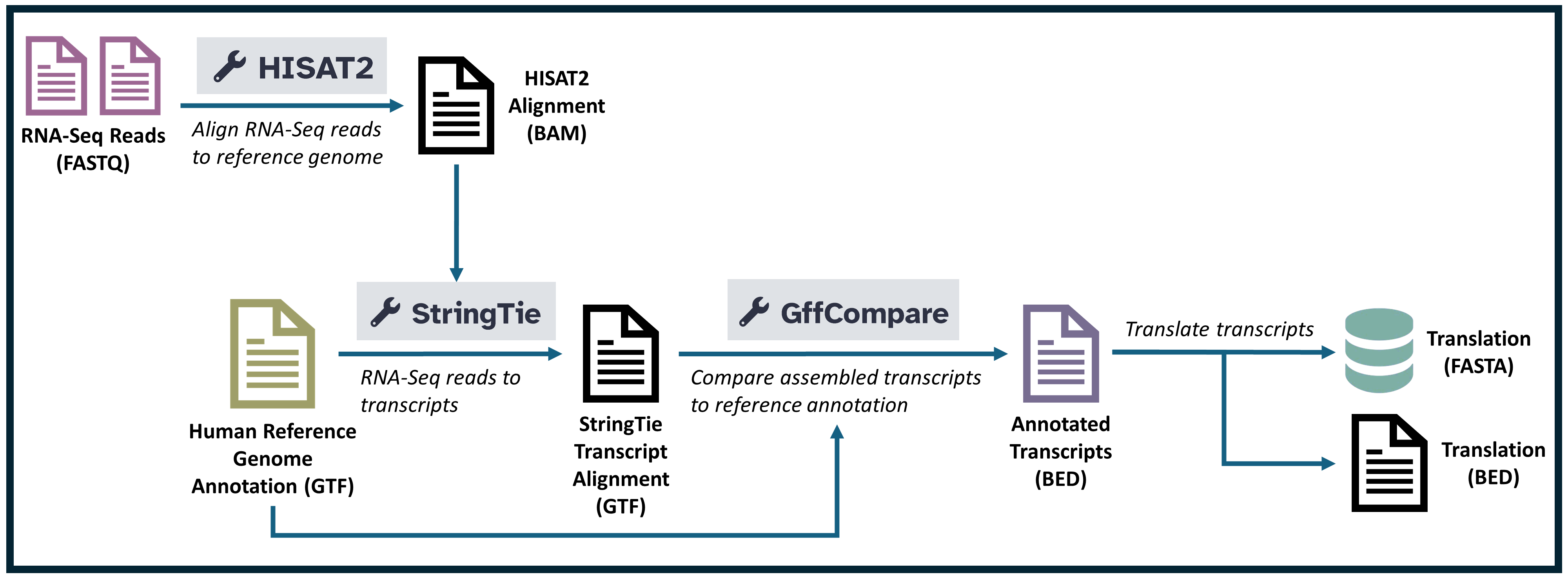
Extracting Assembled sequences with Stringtie and GFF compare
Assemble with StringTie
StringTie is a tool used for assembling RNA-Seq alignments into potential transcripts, estimating their abundances, and creating GTF files of transcript structures. In this task, StringTie takes short reads mapped by HISAT2 as input and uses a reference GTF/GFF3 file to guide the assembly of transcripts. This guidance enables StringTie to produce accurate transcript structures that can be useful for differential expression analysis and downstream functional analyses.
In this workflow, StringTie is responsible for reconstructing transcript structures based on RNA-Seq alignments. By using a reference file to guide the assembly, StringTie improves the accuracy of the transcript annotations, which is valuable for comparing expression levels across conditions or identifying novel transcript variants. This step provides a foundation for gene expression quantification and other transcriptome analyses.
Hands-on: StringTie
StringTie ( Galaxy version 2.2.3+galaxy0) with the following parameters:
“Input options”: Short reads
param-file“Input short mapped reads”: output_alignments (output of HISAT2tool)
“Use a reference file to guide assembly?”: Use reference GTF/GFF3
“Reference file”: Use a file from history
param-file“GTF/GFF3 dataset to guide assembly”: Homo_sapiens.GRCh38_canon.106.gtf (Input dataset)
“Output files for differential expression?”: No additional output
Question
Why is a reference GTF/GFF3 file used to guide the assembly in StringTie?
What type of input does StringTie require to perform transcript assembly?
A reference GTF/GFF3 file provides known transcript information, helping StringTie to assemble transcripts more accurately. This guidance ensures that the assembled transcript structures are consistent with existing gene models, enhancing the reliability of the results and enabling easier comparison across different samples.
StringTie requires mapped short reads as input, typically in BAM format, which contain RNA-Seq alignments. These mapped reads are used to reconstruct transcript structures, and a reference annotation file can be optionally provided to guide the assembly process.
Accuracy of transcript predictions with GffCompare
GffCompare is a tool used to compare and evaluate the accuracy of transcript predictions by comparing GTF/GFF files with reference annotations. In this task, GffCompare takes the transcript assembly output from StringTie and compares it against a reference annotation file. This allows users to identify novel transcripts, assess the accuracy of the assembly, and evaluate the completeness of the transcript predictions relative to known annotations.
In this workflow, GffCompare assesses the quality of the transcript assembly produced by StringTie by aligning it with a known reference annotation. This step helps determine which assembled transcripts match known genes, which are new variants, and which might represent entirely novel transcripts. By providing accuracy metrics and categorizing the transcripts, GffCompare facilitates the interpretation of transcriptomic data, especially in studies aiming to identify new gene isoforms or validate assembled structures.
Hands-on: GffCompare
GffCompare ( Galaxy version 0.12.6+galaxy0) with the following parameters:
param-file“GTF inputs for comparison”: output_gtf (output of StringTietool)
“Use reference annotation”: Yes
“Choose the source for the reference annotation”: History
Why is it important to compare assembled transcripts to a reference annotation?
What information can GffCompare provide about the assembled transcripts?
Comparing assembled transcripts to a reference annotation helps identify which transcripts correspond to known genes, which ones are novel isoforms, and which are new transcript structures. This process validates the transcript assembly and provides confidence in the biological relevance of the findings.
GffCompare provides information on how well the assembled transcripts match the reference annotation, categorizing transcripts as known, novel isoforms, or completely new structures. It also provides accuracy metrics, which are useful for evaluating the quality of the transcript assembly.
BED file conversion with Convert gffCompare annotated GTF to BED
Convert gffCompare Annotated GTF to BED is a tool that converts GTF files annotated by GffCompare into BED format. BED (Browser Extensible Data) files are commonly used for displaying transcript annotations in genome browsers, providing a simpler and more compact representation than GTF files. In this task, the tool converts GffCompare’s annotated GTF output, allowing visualization and further analyses of transcript annotations in a format compatible with many genomic tools and browsers.
In this workflow, converting GffCompare’s annotated GTF output to BED format allows the user to visualize and analyze the assembled and annotated transcripts in genome browsers. This step facilitates the interpretation of transcript locations, boundaries, and structures in an accessible and efficient manner, especially for assessing the genomic context and identifying patterns across the genome.
Hands-on: Convert gffCompare annotated GTF to BED
Convert gffCompare annotated GTF to BED ( Galaxy version 0.2.1) with the following parameters:
param-file“GTF annotated by gffCompare”: transcripts_annotated (output of GffComparetool)
“filter gffCompare class_codes to convert”: ``
Question
Why is BED format useful for viewing transcript annotations?
What does the option “filter gffCompare class_codes to convert” allow you to do?
BED format provides a simplified, compact way to represent transcript locations, which is widely compatible with genome browsers. This format allows easy visualization of transcript structures, making it efficient for examining the genomic context of the assembled transcripts.
The “filter gffCompare class_codes to convert” option lets users specify which types of transcript classes (such as known or novel isoforms) to include in the BED output. This filter is useful for focusing on specific categories of transcripts, such as novel structures, for further analysis or visualization.
Translating BED to FASTA sequences with Translate BED transcripts
Translate BED transcripts is a tool that translates BED files containing transcript annotations into FASTA sequences. This tool uses a reference genomic sequence (in this case, a 2bit file) to extract the nucleotide sequences corresponding to the regions defined in the BED file. The output is a FASTA file, which contains the translated sequences of the annotated transcripts, allowing further analysis of their sequence composition.
In this workflow, translating BED files to FASTA sequences is essential for obtaining the actual nucleotide sequences of the annotated transcripts. This step enables the user to analyze the sequences further, for example, by identifying functional regions, and sequence motifs, or conducting downstream analysis like mutation detection or variant calling.
Hands-on: **Translate BED transcripts**
Translate BED transcripts ( Galaxy version 0.1.0) with the following parameters:
param-file“A BED file with 12 columns”: output (output of Convert gffCompare annotated GTF to BEDtool)
“Source for Genomic Sequence Data”: Locally cached twobit
“Select reference 2bit file”: hg38
In “Fasta ID Options”:
“fasta ID source, e.g. generic”: generic
Question
Why do we use a 2bit reference file in the Translate BED Transcripts tool?
What is the role of the “Fasta ID source” option in this tool?
The 2bit reference file provides a compact and efficient representation of the reference genome sequence, which is used to extract the nucleotide sequences corresponding to the BED annotations. It allows the tool to quickly and accurately retrieve the relevant genomic sequences for translation into FASTA format.
The “Fasta ID source” option specifies the format for the FASTA sequence identifiers. The “generic” option assigns a simple generic identifier to each sequence. This helps in organizing the sequences in a standardized manner, especially if specific identifiers are not available or needed for further analysis.
Generating a genomic coordinate file with bed to protein map
The bed to protein map tool translates the genomic coordinates in a BED file into protein sequences. The input BED file should have 12 columns, where the “thickStart” and “thickEnd” fields define the protein-coding regions. This tool uses these coordinates to map the nucleotide sequences (from the BED regions) into protein sequences, assuming the provided BED file contains protein-coding regions.
This tool is important for converting the genomic annotations (in BED format) that correspond to protein-coding regions into actual protein sequences. This step is critical for downstream protein analysis, such as protein function prediction, domain identification, or understanding the consequences of genetic mutations at the protein level.
Hands-on: bed to protein map
bed to protein map ( Galaxy version 0.2.0) with the following parameters:
param-file“A BED file with 12 columns, thickStart and thickEnd define protein coding region”: translation_bed (output of Translate BED transcriptstool)
Question
What do the “thickStart” and “thickEnd” fields in the BED file represent in the context of protein mapping?
Why is it important to ensure that the BED file contains the correct “protein-coding regions” for this tool to function properly?
The “thickStart” and “thickEnd” fields in the BED file specify the start and end coordinates of the protein-coding regions within the genome. These fields define the portion of the transcript that will be used for translating nucleotide sequences into protein sequences.
The accuracy of the protein sequences produced by the bed-to-protein map tool depends on correctly identifying protein-coding regions in the BED file. If the file includes non-coding regions or incorrectly defined coordinates, the resulting protein sequences may not be accurate, which could lead to erroneous conclusions in downstream analyses.
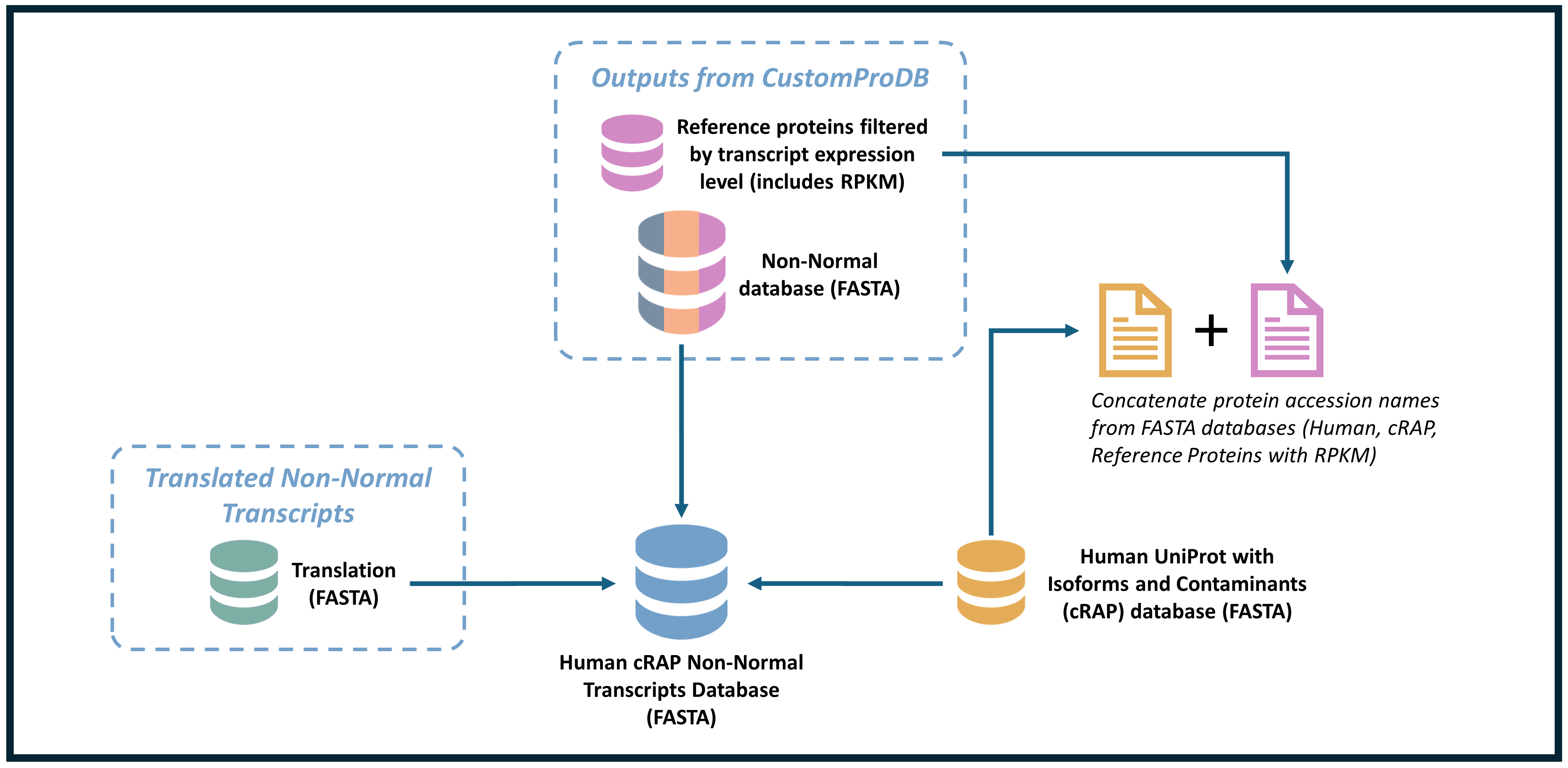
Merging the non-normal databases with the known HUMAN protein sequence
Merging non-normal databases with the known human protein sequence involves integrating data from various sources into a unified format for more efficient analysis. In bioinformatics, this process is often necessary when working with protein sequence data, especially when datasets include variations, unknown sequences, or newly identified proteins alongside well-established reference proteins from the human genome. In this case, we are merging a previously integrated variant database (which includes SNV, INDEL, and RPKM), assembled FASTA data generated from translating BED files to transcripts, the UniProt human reference, and a known contaminant database.
Merging all databases using FASTA Merge Files and Filter Unique Sequences
Hands-on: FASTA Merge Files and Filter Unique Sequences
FASTA Merge Files and Filter Unique Sequences ( Galaxy version 1.2.0) with the following parameters:
“Run in batch mode?”: Merge individual FASTAs (output collection if the input is a collection)
In “Input FASTA File(s)”:
param-repeat“Insert Input FASTA File(s)”
param-file“FASTA File”: HUMAN_CRAP.fasta (Input FASTA database)
In “Input FASTA File(s)”:
param-repeat“Insert Input FASTA File(s)”
param-file“FASTA File”: Non-normal_CustomProDB_FASTA (output of FASTA merge of 3 CustomProDB databasestool)
In “Input FASTA File(s)”:
param-repeat“Insert Input FASTA File(s)”
param-file“FASTA File”: translation_fasta (output of Translate BED transcriptstool)
Conclusion
In this workflow, we demonstrated a comprehensive process for merging non-normal protein sequence data with known human protein sequences, ensuring compatibility and consistency at each step.
Starting with data preparation, we converted sequence data into appropriate formats making it suitable for downstream analysis. Next, we merged non-normal sequences with known human proteins using tools such as FASTA Merge Files, ensuring the datasets aligned based on common identifiers. The subsequent steps, involving Translating BED transcripts and bed to protein map, translated genomic coordinates into protein sequences, further enriching our dataset. This workflow effectively integrates variant/non-normal proteins with established references, offering a robust resource for further analysis in bioinformatics applications like functional annotation and differential expression studies. By combining multiple bioinformatics tools, this process is adaptable to various research needs, making it invaluable for genomic and proteomic analysis. The output from this workflow will be now used for the neoantigen database searching.
Rerunning on your own data
To rerun this entire analysis at once, you can use our workflow. Below we show how to do this:
Click on Workflow on the top menu bar of Galaxy. You will see a list of all your workflows.
Click on galaxy-uploadImport at the top-right of the screen
Paste the following URL into the box labelled “Archived Workflow URL”: https://training.galaxyproject.org/training-material/topics/proteomics/tutorials/neoantigen-2-non-normal-database-generation/workflows/main_workflow.ga
Click the Import workflow button
Below is a short video demonstrating how to import a workflow from GitHub using this procedure:
Video: Importing a workflow from URL
Run Workflowworkflow using the following parameters:
Click on Workflow on the top menu bar of Galaxy. You will see a list of all your workflows.
Click on the workflow-run (Run workflow) button next to your workflow
Configure the workflow as needed
Click the Run Workflow button at the top-right of the screen
You may have to refresh your history to see the queued jobs
Disclaimer
Please note that all the software tools used in this workflow are subject to version updates and changes. As a result, the parameters, functionalities, and outcomes may differ with each new version. Additionally, if the protein sequences are downloaded at different times, the number of sequences may also vary due to updates in the reference databases or tool modifications. We recommend that users verify the specific versions of software tools used to ensure the reproducibility and accuracy of results.
You've Finished the Tutorial
Please also consider filling out the Feedback Form as well!
Key points
Create a customized Variant proteomics database from 16SrRNA results.
Further information, including links to documentation and original publications, regarding the tools, analysis techniques and the interpretation of results described in this tutorial can be found here.
References
Chambers, M. C., P. D. Jagtap, J. E. Johnson, T. McGowan, P. Kumar et al., 2017 An Accessible Proteogenomics Informatics Resource for Cancer Researchers. Cancer Research 77: e43–e46. 10.1158/0008-5472.can-17-0331
Feedback
Did you use this material as an instructor? Feel free to give us feedback on how it went.
Did you use this material as a learner or student? Click the form below to leave feedback.
Hiltemann, Saskia, Rasche, Helena et al., 2023 Galaxy Training: A Powerful Framework for Teaching! PLOS Computational Biology 10.1371/journal.pcbi.1010752
Batut et al., 2018 Community-Driven Data Analysis Training for Biology Cell Systems 10.1016/j.cels.2018.05.012
@misc{proteomics-neoantigen-2-non-normal-database-generation,
author = "Subina Mehta and Katherine Do and James Johnson",
title = "Neoantigen 2: Non-normal-Database-Generation (Galaxy Training Materials)",
year = "",
month = "",
day = "",
url = "\url{https://training.galaxyproject.org/training-material/topics/proteomics/tutorials/neoantigen-2-non-normal-database-generation/tutorial.html}",
note = "[Online; accessed TODAY]"
}
@article{Hiltemann_2023,
doi = {10.1371/journal.pcbi.1010752},
url = {https://doi.org/10.1371%2Fjournal.pcbi.1010752},
year = 2023,
month = {jan},
publisher = {Public Library of Science ({PLoS})},
volume = {19},
number = {1},
pages = {e1010752},
author = {Saskia Hiltemann and Helena Rasche and Simon Gladman and Hans-Rudolf Hotz and Delphine Larivi{\`{e}}re and Daniel Blankenberg and Pratik D. Jagtap and Thomas Wollmann and Anthony Bretaudeau and Nadia Gou{\'{e}} and Timothy J. Griffin and Coline Royaux and Yvan Le Bras and Subina Mehta and Anna Syme and Frederik Coppens and Bert Droesbeke and Nicola Soranzo and Wendi Bacon and Fotis Psomopoulos and Crist{\'{o}}bal Gallardo-Alba and John Davis and Melanie Christine Föll and Matthias Fahrner and Maria A. Doyle and Beatriz Serrano-Solano and Anne Claire Fouilloux and Peter van Heusden and Wolfgang Maier and Dave Clements and Florian Heyl and Björn Grüning and B{\'{e}}r{\'{e}}nice Batut and},
editor = {Francis Ouellette},
title = {Galaxy Training: A powerful framework for teaching!},
journal = {PLoS Comput Biol}
}
Congratulations on successfully completing this tutorial!
Go Further
Do you want to extend your knowledge? Follow one of our recommended follow-up trainings:
Questions: